
Acid Cleaning Coverslips
- Make-up 300 ml of 2 parts Nitric Acid – 1 part HCl in a glass beaker in the hood. Solution will turn orange-red.
- Place #1.5 coverslips into solution a few at a time. Let sit for about 2 hrs. with occasional swirl.
- Decant acid into waste bottle carefully.
- Wash extensively in running water until pH of water is back up to 5.5 -6.0.
- Store in 70% EtOH.
- Flame coverslip in hood prior to use.
Fluorescence Mounting Medium (Antifade)
Materials Needed
- 20ml glass scintillation vial
- Small stir bar
- Foil
- Glycerol
- 1X PBS
- Pipets
- * P-phenylenediamine ( EMD Chemicals Inc. Cat# PX0730)
- Carbonate-Bicarbonate Buffer (see below)
- Wrap a glass scintillation vial with foil and drop in a small stir bar. (PPD is light-sensitive.)
- With a 10 ml pipet add 9 mls of glycerol to the vial.
- With the 1000 ml Pipetman add 1ml of 1X PBS.
- Place on stirrer and begin mixing.
- Weigh out 10 mg of p-phenylenediamine on the Mettler balance.
PPD is toxic. Wear gloves and don’t inhale it. - Add the PPD to the vial and stir untill it is all in solution (1-2 hrs.). The medium should appear almost colorless to a slight tint of yellow. If it is an intense yellow or orange color the PPD is most likely contaminated and will have background staining.
- pH the mounting medium to a pH of 8.0-9.0 using the Carbonate-Bicarbonate buffer. pH paper of range 6.5-10.0 should be used to check the pH of the medium after addition of 12 drops of the Carb-Bicarb buffer and stirring. Additional drops of buffer are added untill the desired pH is reached.
- Aliquot the mounting medium and store at -70oC.
* Flakes of PPD are large and should be crushed
Carbonate- Bicarbonate Buffer
- Make up a 0.2M solution of anhydrous sodium carbonate (2.12g/100ml)
- Make up a0.2M solution of sodium bicarbonate (1.68g/100ml)
Take 4 mls of A + 46 mls of B and bring up to 200 mls with DH2O. The pH will be 9.2.
*Note: If the PPD is contaminated or goes bad ( turns a brown color ) it will stain DNA, so each preparation should be tested. Check by looking at mitotic cells to be sure that chromosomes are not stained.
Immunofluroescence Technique
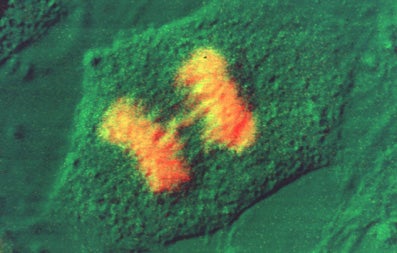
- Fix cells in 2% formaldehyde in PBS/pH 7.4 for 15 min. at 20oC. 2% formaldehyde is made up fresh prior to use by dissolving the appropriate amount of EM grade paraformaldehyde (Prill form, from Electron Microscopy Sciences) in PBS and heating on a hot plate in the hood (setting of 5-6) until the aldehyde goes into solution. Keep the bottle cap loosened so that pressure does not build up. Cool down to 20oC and pH to 7.4. Alternatively, cells may be fixed in 100% methanol at -20oC for 3 minutes. If methanol fixation is used skip to step 4.
- Wash in PBS 3 X 10 min.
- Permeabilize in 0.2% Triton X-100 plus 1% normal goat serum (NGS) in PBS/pH 7.3 for 5 minutes on ice.
- Wash in PBS + 1% NGS 3 X 10 min.
- Incubate in the appropriate concentration of primary antibody for 1 hour at room temp. in a humidified chamber. If using 22mm X 22mm square coverslips, 30 ul of diluted antibody is placed on the coverslip and the coverslip is inverted onto a glass slide. The slide is then placed in the humidified chamber which is incubated at room temp.
- Wash in PBS + 1% NGS 3 X 10 min.
- Incubate in secondary antibody (FITC or Texas Red conjugated) at a dilution of 1:50 for 1 hour in a humidified chamber at room temp.
- Wash in PBS 4 X 10 min.
- Mount coverslip with a drop of mounting medium and seal coverslip with clear nail polish to prevent drying and movement under the microscope.
From: Spector, D.L. and H.C. Smith. 1986. Exp. Cell Res. 163, 87-94.
Post-Embedding Immunogold Method
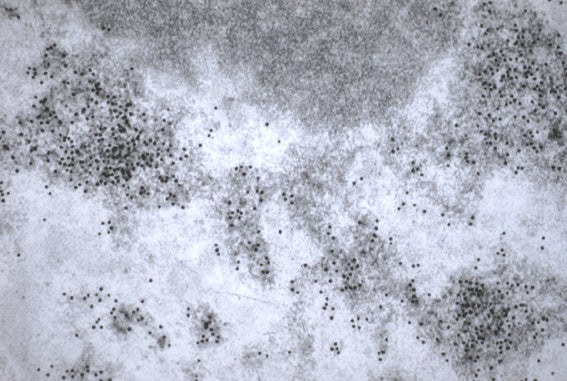
- L.R. white sections are collected on gold Veco grids.
- If blocking is necessary for nonspecific labeling, buffer is supplemented with normal goat serum (2 – 10%) and bovine serum albumin (0.2 – 1%). We usually start with a 2% NGS and 0.2% BSA block and increase if necessary up to 10% and 1% respectively. Grids are floated face down on drops of blocking solution in tris buffered saline with 1.0% tween-20 and incubated for 60 min.
- Grids are then transferred to drops of appropriately diluted primary antibody in tris-buffered saline, pH 7.6, with 1.0% tween-20. Grids are incubated in the refrigerator, approx. 4oC, overnight in a humidified chamber.
- The following morning, grids and solutions are allowed to come to room temperature.
- Grids are washed 10 X 1 min. each (or longer and more frequently, if needed) in buffer.
- Transfer grids to appropriate gold-labeled secondary antibody (1:20) diluted in tris- buffered saline with 1.0% tween-20. First spin diluted gold conjugated antibody in a microfuge at middle speed for 30 seconds – 1 minute to get rid of gold clumps. Incubate for 60 mins. at room temperature.
- Wash grids 10 X 1 min. each in buffer.
- Wash grids 10 X 1 min. each in double-distilled H2O.
- Counter stain (if desired) for E.M. with UA/Pb.
Tris-buffered saline, pH 7.6
0.02M tris
0.15M sodium chloride
1.0% tween-20
pH to 7.6
From: Spector et al., 1991. EMBO J. 10, 3467-3481.
mRNA In Situ Hybridization with Nick-Translated Probes
- Cells are fixed with freshly made 4% formaldehyde in PBS, pH 7.4 for 15 min at room temperature. All solutions should be made in Molecular Biology grade ultrapure water (no RNase). Wear gloves at all times and use sterile disposable pipets and tips.
- After rinsing in PBS (3 X 10 min. each), cells are permeabilized with 0.5% Triton X-100 in 1X PBS for 10 min at 4oC.
- Cells are then rinsed in PBS (3 X 10 min. each) and then in 2X SSC (1 X 5 min.).
- 100 ng of nick translated probe (containing digoxigenin dUTP) and 20 ug of competitor E. coli tRNA per coverslip are dried down in a Speed Vac (Savant). This is a good starting place but you may have to titrate your specific probe.
- 10 μl of deionized formamide is added to the dried DNA.
- The probe and tRNA are denatured by heating for 10 min at 90oC. The probe is chilled on ice immediately.
- 10 μl of Hybridization buffer (20% dextran sulfate + 4X SSC) is added to the denatured probe so that the final concentrations in the hybridization mixture are 5 ng/ml of probe, 1 ug/ml of E. coli tRNA, 2X SSC, and 10% dextran sulfate.
- 20 μl of hybridization mixture/probe is placed onto each coverslip.
- Coverslips are inverted onto a slide and sealed with rubber cement and incubated in a humid chamber for 16 hrs. at 37oC.
- After rinsing in 2X SSC/50% formamide at 37oC, 2X SSC and 1X SSC at room temperature for 30 min. each, the coverslip containing cells are incubated in 4X SSC/0.25% BSA/2ug/ml anti-digoxigenin antibody for 60 min. in a humid chamber at room temperature in the dark.
- Coverslips are then rinsed in 4X SSC (1 X 15 min.) at room temperature, 4X SSC/0.1% Triton X-100 (1 X 15 min.), and 4X SSC (3 X 10 min. each).
- Coverslips are mounted in fluorescence mounting medium.
Modified from: Jim~nez-Garcia, L. and D.L. Spector. 1993. Cell 73, 47-59.
DISCLAIMER: The linked protocols are those that we routinely use in the laboratory. Some of the procedures and chemicals are extremely dangerous and therefore we assume that the protocols will only be used by competent laboratory staff. We assume no responsibility for any problems encountered, either scientific or personal, due to the use of these protocols.